Tools

20/01/2022
Thesis defence of Adrià Pérez: "Learning How to Simulate: applying machine learning methods to improve molecular dynamics simulations"
Next Thursday 27th of January at 10:00 am, Adrià Pérez, member of the Computational Science group of GRIB will read his thesis: “Learning How to Simulate: applying machine learning methods to improve molecular dynamics simulations”.
The event will be online. Free access at https://upf-edu.zoom.us/j/89823439190
Abstract:
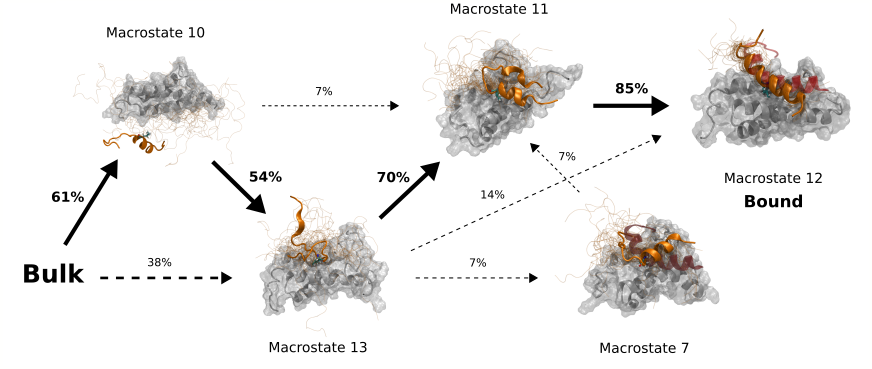
Characterizing protein dynamics is critical to understand the connection between sequence and function. Molecular dynamics simulations are one of the predominant techniques to study protein dynamics due to their capacity to capture dynamical processes of proteins across different timescales with atomic resolution. However, molecular dynamics has limitations that so far limited the ability to become a surrogate model of real protein dynamics, mainly sampling limitations due to the high computational cost and force field accuracy.
In this thesis we tackle these issues with the latest advances in machine learning. In the first part of this thesis, we will develop a novel adaptive sampling algorithm inspired by reinforcement learning methods, and we will later apply it to reconstruct the full binding event between an intrinsically disordered protein and its binding partner. In the second part of this thesis, we develop TorchMD, a deep learning framework for molecular simulations and apply it to learn a coarse-grained potential for protein folding simulations.