Tools

7/6/2011
A major step in molecular simulation - key to designing new drugs
The research has been published online in the prestigious magazine Proceedings of the National Academy of Sciences (PNAS)
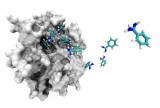
Researchers of GRIB (IMIM/ UPF) have successfully reproduced and reconstructed the complete process of a small molecule binding with its target protein. This advance enables the drug's affinity and binding level with the protein to be calculated as well as understanding the interactions established by the drug in order to act, thus moving towards safer and more efficient design of new drugs.
This groundbreaking project helps show a process that was hitherto invisible and therefore unknown, and opens up a new avenue in the design of new drugs.
The process of a drug, usually a small molecule, binding to its target protein is highly dynamic and depends on interactions at a nanometric scale (billions of times smaller than a metre) and occurs at nano timescales - in microseconds (billions of times faster than a second).
The capturing of movements of small molecules with a resolution of up to an atom is beyond current technical capabilities. However, using computer techniques, it is possible to represent the molecules at atomic scale and reproduce their movements with high mathematical precision. Understanding how the protein binds to a molecule - in which the molecule is capable of being recognised by another, causing a biological response (binding) - is vitally important for the design of new drugs. Despite the progress made so far with the technique, no study had provided a complete reconstruction of the protein-ligand binding process. "The method provides not only the binding affinity and the kinetics of the reaction, but also information about the atomic resolution during the process: binding sites, transition states and metastable states are potentially useful for expanding the probability of success when designing drugs. This methodology can be directly applied to other molecular systems and is therefore of general interest in biomedical and pharmaceutical research" explains Gianni de Fabritiis, coordinator of the Computational Biophysics Laboratory of the Biomedical Informatics Research Programme (GRIB- IMIM/ UPF).
The researchers are now working to expand the applicability of this methodology and make better use of the computational capabilities as, in cases in which ligands are larger and more flexible and where the proteins involve more complex binding processes, greater computational effort is required.
Reference article:"Complete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulations" I Buch, T Giorgino, G De Fabritiis.